
EAEU legislation for Medical Device registration has been continuously updating for the last six years since the Eurasian Economic Union, otherwise known as EAEU, was entered into force in 2015. Five countries have signed the “Treaty on the Eurasian Economic Union” to create a single regional market and ensure free movement of products and services while fostering stable development. Those five countries consist of Belarus, Kazakhstan, Russia, Armenia, and Kyrgyzstan.
Up until 2021, companies could register their medical devices in the member states through two pathways:
- National systems;
- New regional EAEU registration system, available since 2017.
Most organizations have continued to register medical devices through national systems. However, as the transition period ends at the end of 2021, all new medical device registrations will have to follow the new regional EAEU registration procedure, starting January 1st, 2022.
In this article, we will review the outline of the must-know legislation for medical device registration in the EAEU:
Understanding the EAEU Legislation for Medical Devices
EAEU Regulations Hierarchy
EAEU legislation describes its law hierarchy and how the regional union functions in its daily activities. The highest authority is the World Trade Organization, whose agreements function as the basis of the economic structure and economic relationships in the Eurasian Economic Union. These are followed by EAEU legislation, with the primary regulation remaining the EAEU Treaty.
The national regulations fall at the bottom of the structure and are effective only in areas not regulated by the EAEU laws. For example, medical device samples’ importation for testing in Russia must be performed following the Russian national procedures. However, such aspects of regulations may change once the EAEU registration system fully kicks off in 2022.

EAEU Legislation Structure
The central law governing medical devices in the Eurasian Economic Union is the Agreement of Common Principles and Rules for Circulation of Medical Products (Medical Devices and Medical Equipment). As you can see, medical products are defined separately as medical devices and medical equipment instead of one definition. This separation is created on purpose to harmonize the legislation as definitions vary country by country. For example, only medical device definition exists in Russia without additional definitions for medical products, equipment or appliances. The new regional EAEU system will erase these discrepancies to ensure smooth flow and execution of the registration procedures.
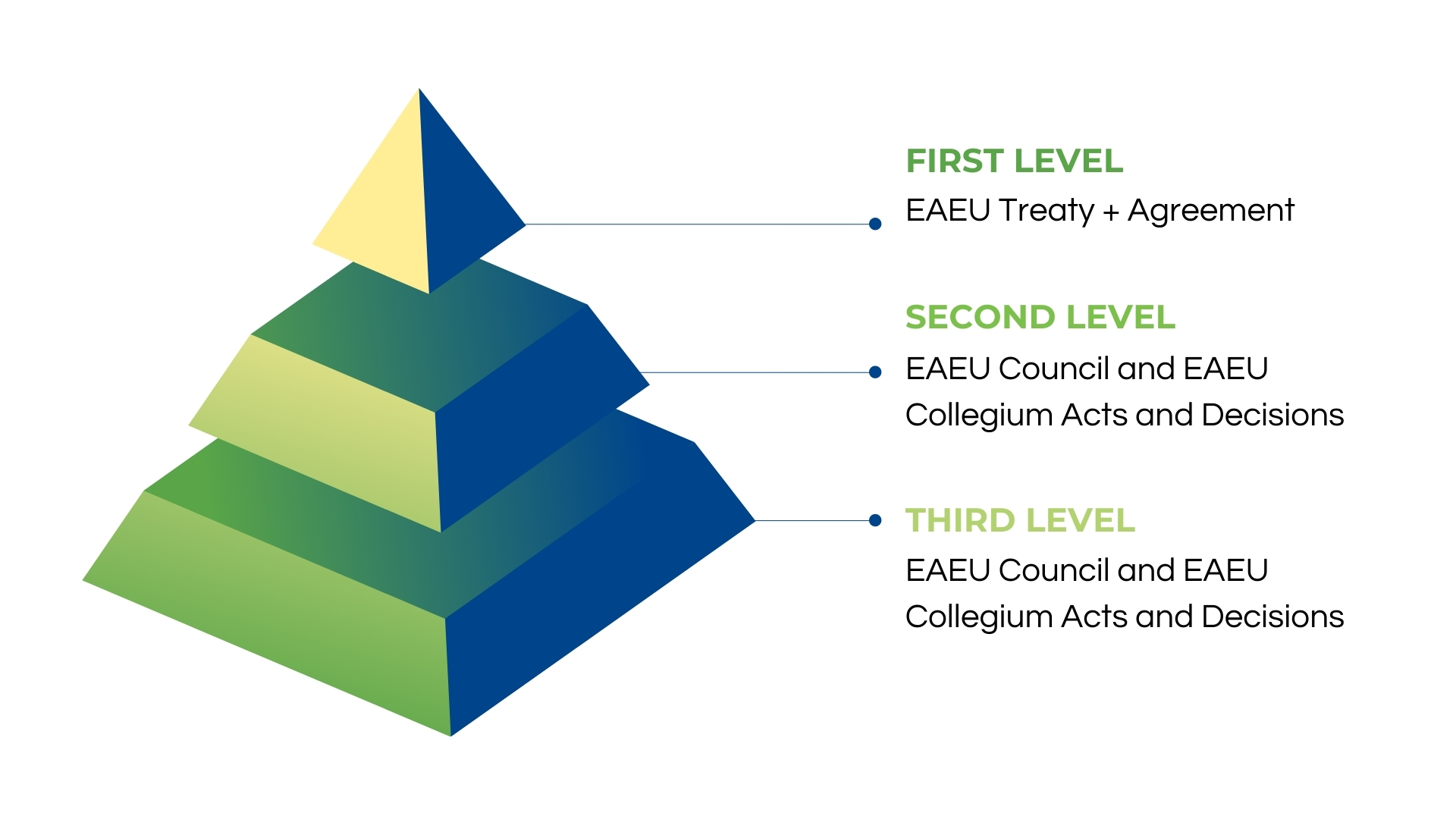
Most regulations are created at the second level as EAEU Council and Collegium Acts and Decisions. Yet, there is a lack of recommendations due to the novelty of the procedure itself. Most explanations of the mentioned rules can be gathered from national expert authorities verbally during seminars, but the interpretations differ. Moreover, the same experts have different views towards the same laws raising more questions than providing answers.
In total, there are about fifty different laws as single regulations instead of one comprehensive document. Around thirty of these rules have a direct impact on the registration procedure. In addition, the entirety of different documents causes discrepancies in described approaches, duplicates of terminology and differences in definitions triggering more challenges down the road.
Main EAEU Legislation Regulations
As mentioned, about fifty distinct rules are regulating medical device registration in the Eurasian Economic Union. We have highlighted the sixteen most important documents that are must-know to understand the new registration system better. You can find them here: Basic EAEU legislation.
Medical Device Regulations: EAEU vs. Russia
What Is a Medical Device? Defining Medical Products.
Medical devices in Russia and EAEU are classified differently, with the central part of the definition overlapping. The essential difference in the definitions is the drug support for medical devices:
- In Russian legislation, medical devices are those “which function is not implemented by pharmacological, immunological, genetic or metabolic effects on the human body.”
- In EAEU regulations, medical devices are those, which cannot be realized by pharmacological, immunological, genetic or metabolic effects on the human body, however, can be supported by drugs.”
A significant update regarding drug support for medical devices is finally described in the region’s laws, clarifying and harmonizing the current legislation.
What Are the Overlaps and Differences in Medical Device Legislation?
Many discrepancies are noticed in the medical device regulations when comparing them to the national registration procedures. Comparing EAEU and Russian legislation, there are five main differences:
- Territory of circulation
- Authorized Representatives of the Manufacturer responsibility
- Operational documentation language
- Document approval
- Approach to quality, effectiveness, and safety confirmation.
With Russian national registration, a medical device can circulate only in the Russian Federation, while the Authorized Representative of the Manufacturer is responsible only for the mentioned circulation. However, if a medical device is registered under EAEU legislation, it can circulate in all EAEU member states, while the Authorized Representative is responsible for all member states as well. In this case, the operation documents must be translated to all EAEU member states’ languages, not only Russian.
In addition, under Russian legislation, documents are approved by the Manufacturer, but under EAEU regulations, they can be approved by the Applicant instead. Nevertheless, the most crucial difference is number five, describing a new approach to quality, effectiveness and safety confirmation.
What Are the Changes in a Medical Device Registration Dossier?
In current national Russian legislation, the medical device registration dossier contains the application document and approximately eleven additional documents, from technical testing reports to expert conclusion reports for stage 1 and 2. However, in EAEU legislation, a second application for the EAEU expertise and approximately thirty additional documents are necessary for the submission.
Furthermore, new types of documents are required, such as manufacturing plant inspection report, post-marketing surveillance plan, and expert conclusion of the reference country.
| NATIONAL RUSSIAN LEGISLATION | EAEU LEGISLATION |
| 1. Application for the state registration and approx. 11 additional documents, including: |
1. Application for the EAEU registration 2. Application for the EAEU expertise, and approx. 30 additional documents, including: |
| 2.Technical testing Report | 3. Technical testing Report |
| 3. Toxicology testing Report | 4. Toxicology testing Report |
| 4. Expert Conclusion I stage | 5. Clinical Trials Report |
| 5. Permission for Clinical evaluation | 6. Manufacturing Plant Inspection Report |
| 6. Clinical evaluation Report | 7. Post-marketing surveillance Plan |
| 7. Expert Conclusion II stage | 8. Expert Conclusion of Reference Country |
Risk Classification: What Are the Differences between Russia, EAEU and EU?
The classification of medical devices into risk classes is the same in Russia and Eurasian Economic Union. Still, it differs significantly from the European Union classification regarding the first risk class. EU has a more detailed categorization of low-risk medical devices, while EAEU is going in another direction with the generalization of these medical products. You can find the detailed comparison between Russia, EAEU and EU in the table below:
| RISK CLASS | EU MDR CLASSIFICATION | EAEU CLASSIFICATION | NATIONAL RU CLASSIFICATION |
| High risk medical devices | Class III | Class III | Class III |
| Medium/High risk medical devices | Class IIb | Class IIb | Class IIb |
| Medium risk medical devices | Class IIa | Class IIa | Class IIa |
| Reusable surgical instruments | Class Ir | Class l | Class l |
| Low risk devices with measuring function | Class Im | ||
| Low risk sterile devices | Class Is | ||
| Low risk non-sterile devices | Class I |
New Approach to General Safety and Performance Requirements (GSPR): Ensuring Quality, Efficacy, and Safety
Eurasian Economic Union has presented a new approach to general safety and performance requirements, otherwise known as GSPR, to ensure the quality, efficacy and safety of medical devices. The approach is the same as EU MDR regulation: “medical devices shall achieve the performance intended by the manufacturer and shall be designed and manufacturing in such a way that during normal conditions of use they are suitable for their intended purpose”. This remains the cornerstone of general safety and performance requirements in the EAEU legislation as well.
Transition Period
On March 9th, 2021, the Eurasian Economic Union commission issued an amendment to the Agreement on Common Principles and Rules for the circulation of Medical Products. It depicts the validity of national marketing authorizations: all national marketing authorizations are valid until their expiry date in the respective member states. However, after expiry, all medical devices will have to be re-registered under the new EAEU legislation.
In addition, authorities will accept applications for national registration until December 31st, 2021. If the applicant submits an application before this date, the registration can be carried out under national member state regulations in 2022. Any submissions after the end of this year will have to follow the Eurasian Economic Union procedure without exceptions.
Medical Device Dossier in the EAEU
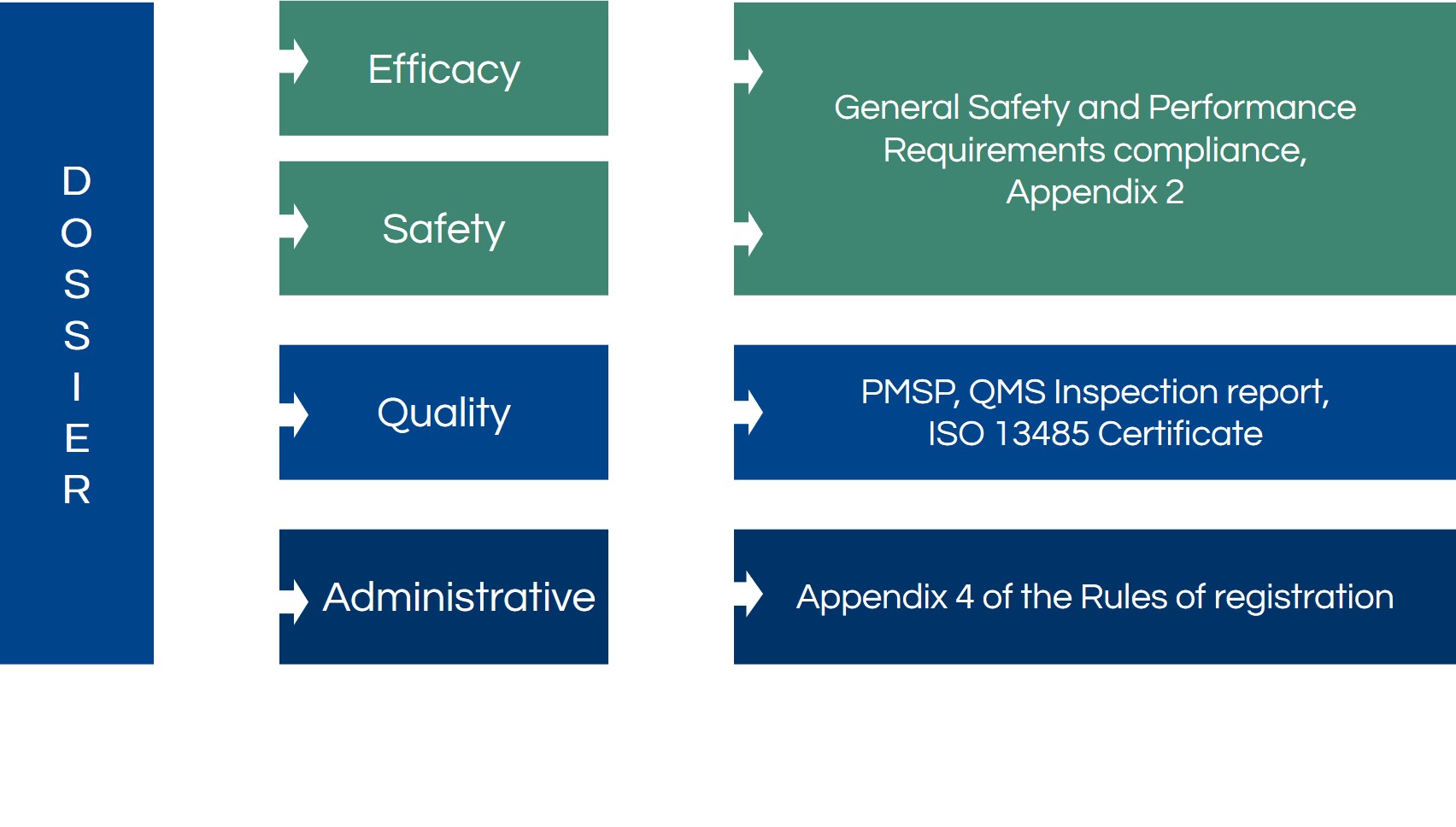
Medical Device Dossier’s Structure
The general dossier structure consists of four parts:
- Efficacy
- Safety
- Quality
- Administrative
The efficacy and safety of the medical device should be presented through general safety and performance requirements and their comprehensive descriptions in the mentioned document. However, 2a, 2b, and 3rd class medical devices’ quality should be confirmed by the quality management system or QMS and post-marketing surveillance report.
How to Prepare the Registration Dossier by the New Regulations?
To prepare the dossier, the applicant must take three main steps:
- Step 1. The applicant should receive preliminary consultations from an expert organization on the registration and examination of a medical device. This step is not mandatory and is applicable only if necessary. However, as most registrations have been completed through national systems until this day, the actual situation will be seen only after the start of 2022, when the EAEU registration procedure will start in full force.
- Step 2. The applicant should receive preliminary consultations from an expert organization on the registration and examination of a medical device. This step is not mandatory and is applicable only if necessary. However, as most registrations have been completed through national systems until this day, the actual situation will be seen only after the start of 2022, when the EAEU registration procedure will start in full force.
- Step 3. Finally, the applicant must fill an application to receive permission to start clinical trials in EAEU and, after conducting the study, include the data in the registration dossier.
The Eurasian Economic Union’s medical device registration system is still adjusting and updating its recommendations: the details and small regulations are continuously changing as there are some discrepancies and gaps that can cause future issues. However, as the new system is coming into effect starting the year 2022, the essential must-know points for medical device legislation in the EAEU remain the same: what is the EAEU medical device legislation structure? What are the new regulations and approaches? What are the differences in Russian, EAEU and EU medical device regulations? What is the transition period, and what happens after it expires?



")

")
")
