Since the establishment of the Eurasian Economic Union in 2015, a significant change is quickly coming into life: a new regional registration system for medical devices is coming into effect from the start of 2022. As most registrations are completed through national procedures, the upcoming regional process raises many questions. Thus, in this article, we will review the medical device registration path in the Eurasian Economic Union:
Conformity Assessment Routes for Medical Device Registration in the EAEU
Stages of the Registration Process in the EAEU
The registration process in the Eurasian Economic Union consists of two main stages:
- Pre-submission
- Post-submission
The pre-submission stage is the registration dossier’s preparation. This step comprises preparing the dossier’s administrative part, performance of technical testing, biological studies, and clinical trials. The fill list of documents can be found in appendix four of the Council Decision No. 46.
The post-submission stage describes the review of the registration dossier. After the dossier is submitted to the Competent Authority of the reference country, it must be reviewed by it as well. If needed, manufacturing plan inspections will be concluded. However, it does not apply to all medical devices, only for 2a sterile, 2b and 3 Risk Classes. After the positive expert conclusion, the medical device can be mutually recognized by the member states and registered in the EAEU region.
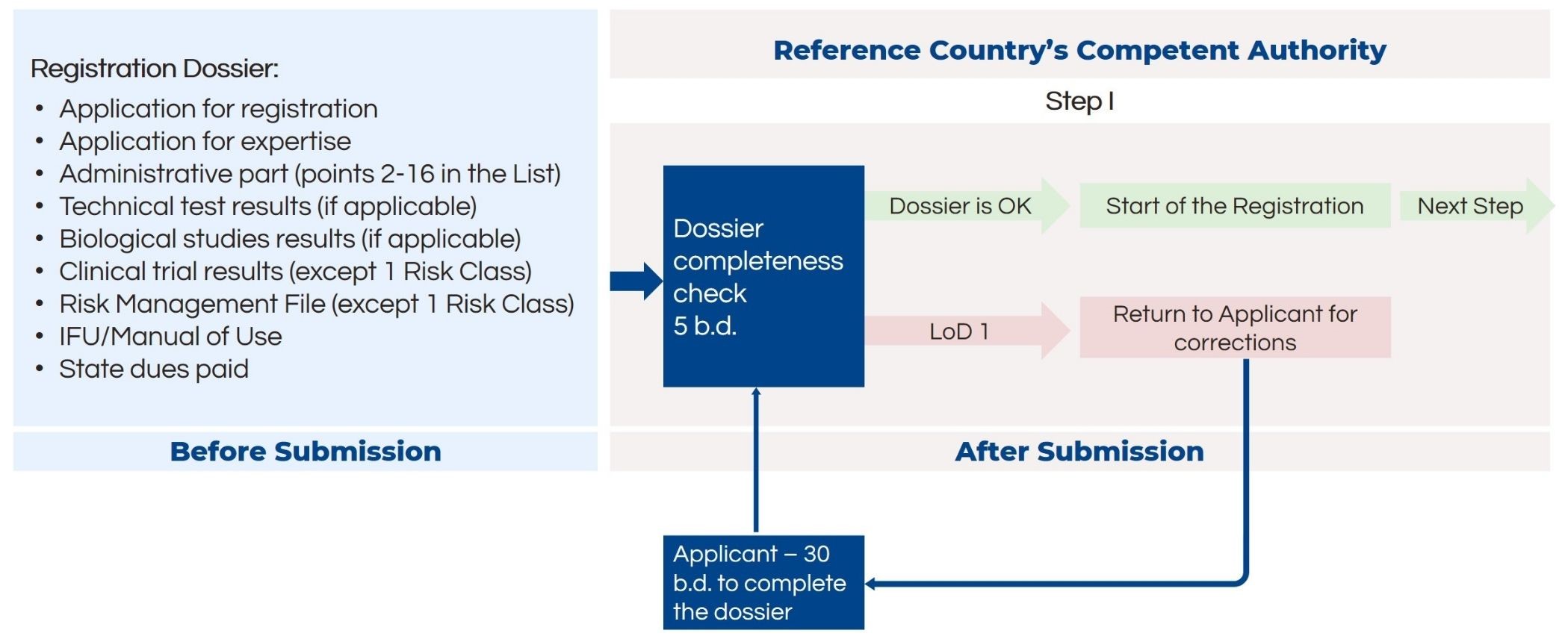
The first step after submission is a formal check of the dossier completeness and application correctness. If additional corrections are needed, the application is sent back to the applicant for corrections.
Risk Classes and Conformity Assessment Routes
Medical Devices in Risk Classes 1 and 2a
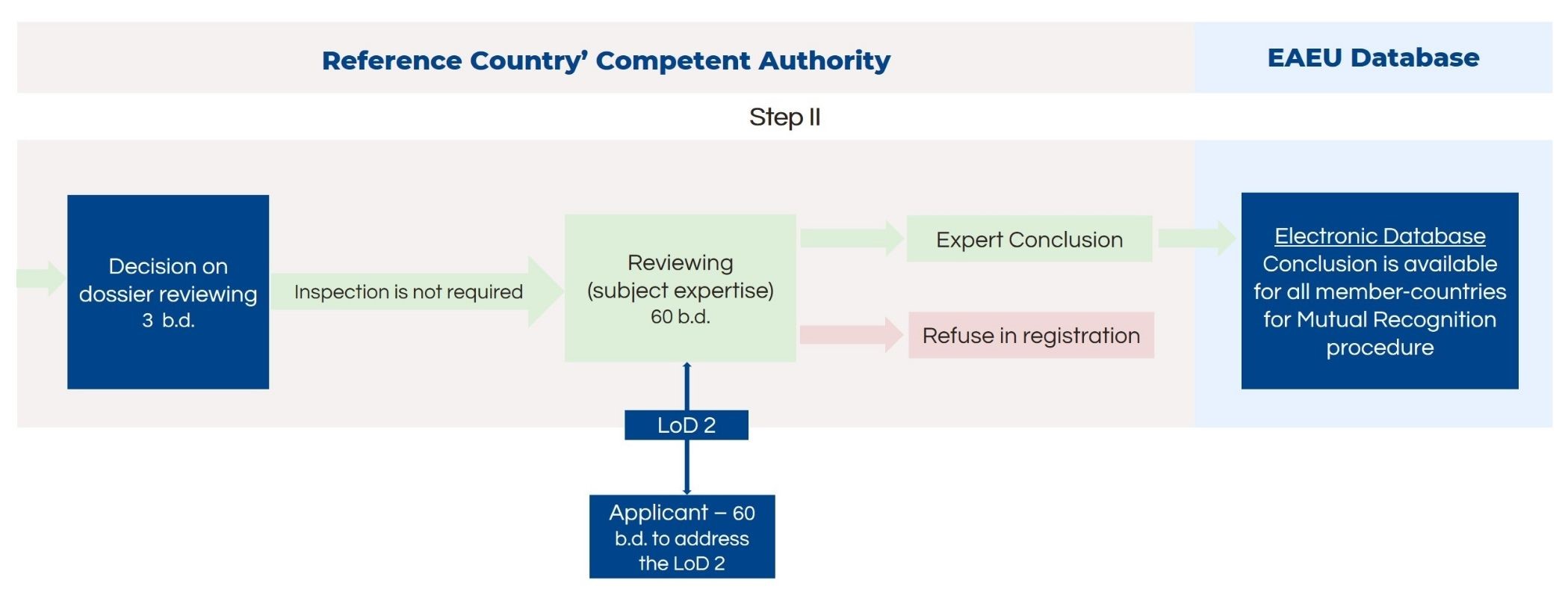
For medical devices in risk classes 1 and 2a, the manufacturing plant inspection is not needed. Thus, after the formal dossier check, the next step – subject expertise review – is initiated. The inspection lasts up to 60 business days. In case the authority finds deficiencies in the dossier, it will inform the applicant with a letter of deficiency. The applicant has 60 days to respond to the authority. If the responses are satisfactory, the authority will pass along the expert conclusion to all member states for mutual recognition.
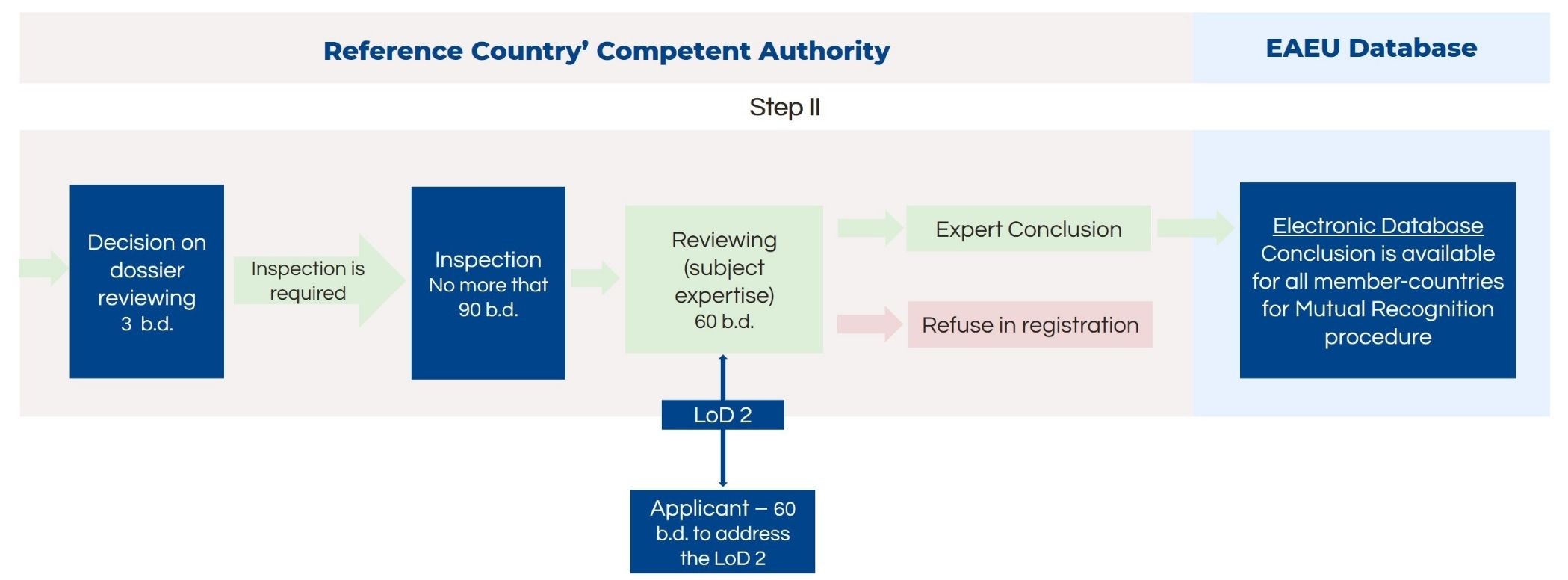
Medical Devices in Risk Classes 2a sterile, 2b and 3
The conformity assessment route for medical devices in risk classes 2a, 2b and 3 is very similar, except it has an additional step – quality management system inspection of the manufacturing site before the expert review and conclusion.
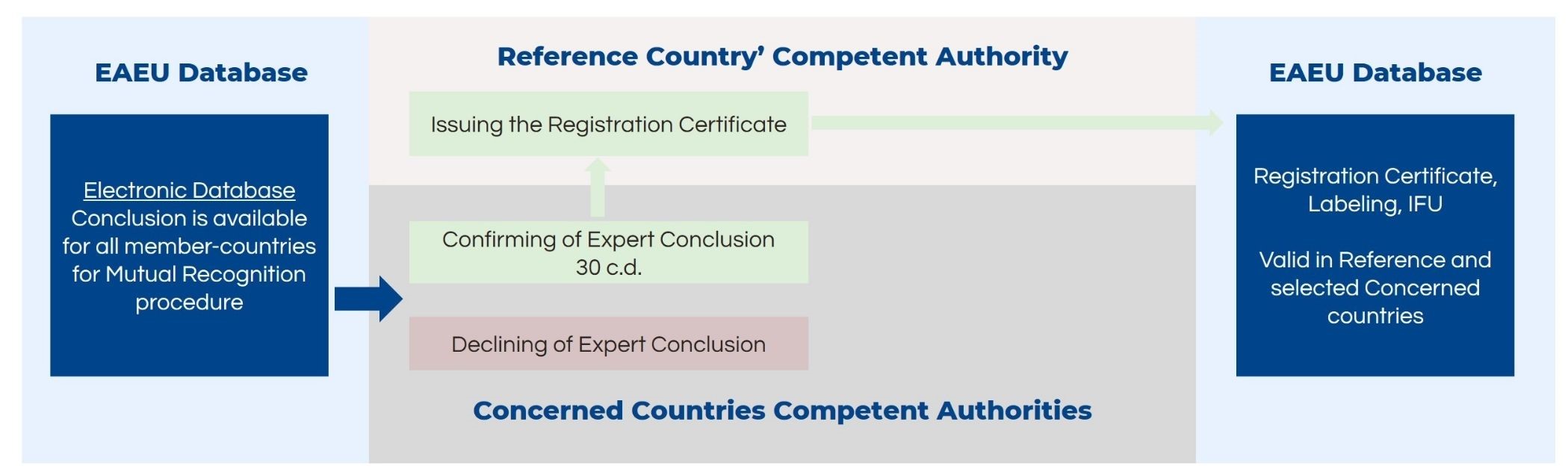
Completion of the Medical Device Registration in the EAEU: Mutual Recognition
The Competent Authorities of the concerned member states will either confirm the expert conclusion for mutual recognition or refuse the recognition within 30 days. If the Competent Authorities do not provide any comments within the designated time, it is considered to be an approval without any objections, and the registration can be finalized. The reference country will issue the registration certificate where all member states are included.
Importation of the Samples for Testing
Currently, the samples for testing can be imported to the Eurasian Economic Union under national regulations. The application can be submitted via the electronic portal – you must receive specific permission before starting the importation procedure.
Technical Testing
Technical testing in the Eurasian Economic Union is mandatory for all medical devices falling into risk classes 1, 2a, 2b and 3. However, IVD products are exempt from this regulation. It is allowed to perform field testing for capital equipment without importation, e.g., tomography unit. All testing must be performed only in the duly authorized testing labs.
The essential documents that must be provided consist of specification, principal explanation, testing protocol, labelling and info packaging and national test reports.
It is important to remember that the national and international standards for technical testing in the EAEU are being updated, and new standards are replacing the old ones. While preparing for technical testing, it is essential to review any recent changes.
Biological Testing
Biological testing is applicable for all medical devices that come into contact with the patient. However, IVDs are exempt from biological testing as well. Biological evaluation includes physical and chemical testing and biological evaluation in viva and in vitro.
Biological testing includes the following stages:
- Safety data analysis
- Reviewing of the documentation and biocompatibility test reports
- Operational documentation analysis
- Sampling and samples identification
- Estimation of the contact duration
- Biological testing itself
- Test reports issuing
Biological testing must be performed in duly authorized labs, just as technical testing.
Clinical Trials in EAEU
The most complex part of the registration procedure in the Eurasian Economic Union is the clinical trials. Clinical trials are applicable to all medical devices except IVDs. For the latter, there are special studies called Clinical Laboratory Trials.
For medical devices with a higher risk factor, i.e., in risk classes 2b and 3 (implantable medical devices), multi-central trials should be conducted. In detail, it means the clinical studies must be done in several countries in the EAEU region instead of one.
Permission for clinical trials is required for all devices except IVDs – only a notification is needed. After the competent authority of the reference country permits the studies, they can be conducted only in duly authorized clinical. It is necessary to remember that the technical file must be provided, as clinical trials cannot be executed without it.
Due to the lack of practical experience with Clinical trials of 2a sterile, 2b and 3 risk classes, there are many vague points for both Authorities and Applicants. An often question is if the mandatory local clinical trials may be conducted in parallel with the technical assessment before submission. Due to the vague regulation, we can only try to theoretically apply the national rules, meaning that first we should perform biology studies, after that – technical testing, and only then the clinical trials. It means that these processes cannot be parallel.
Manufacturing Plant Inspection
First of all, the manufacturer should implement and maintain the quality management system for medical device manufacturing. The authority inspects the manufacturing plant before the preparation for the expert conclusion. The timelines for inspections are not included in the registration timelines but should not exceed 90 days.
Subject Expertise
The first six steps of subject expertise include:
- Analysis of documents and materials determining safety, efficacy and quality of the medical product.
- Analysis of data on the development and manufacture of the medical product.
- Analysis of the standards to which the medical product corresponds.
- Analysis of the technical testing protocols and results.
- Analysis of reports on the results of an inspection of the medical product manufacturer.
- Analysis of reports on the evaluation of the biological action of the medical product and results.
There are many steps in the comprehensive analysis of the application, and it’s worth highlighting that the risk management plan must be included in full.
If your medical device contains a drug product, you must provide all the necessary data on drug safety. Furthermore, if your medical device is software, you must provide all of the documents for software validation.
When preparing for the application, you should include the post-market surveillance plan describing the data collection plan for safety and efficacy measures after potential marketing authorization.
EAEU Mutual Recognition Procedure
After the expert opinion is available, the reference state’s authority places its conclusion on the information system. This should be done in 30 days. However, as mentioned before, if they do not provide the opinion in the specified time, it is considered an approval without objections.
However, there might be conflicts between the authorized authority of the reference country and the concerned member states. In a case of non-consensus, a third party – a medical advisory committee – is involved, and they will review all the materials and results.
There are some limits in approval procedure when there is no consensus: “Non-approval of the expert opinion of the reference state in one of the states concerned is the basis for refusal to use the medical product in the territory of that state”. For example, if you choose the Russian Federation as a concerned country and Belarus as a reference country, and the Belarus authority declined the application, then the registration certificate could be issued only for the Russian Federation. This would allow you to market your products only in a limited territory of the region.
—
The new registration system in the Eurasian Economic Union is coming into full effect next year, but with many vague regulations, it raises more questions than provides answers. A new approach to clinical trials, testing process, different conformity assessment routes for medical devices with varying risk levels are just part of the practical implications coming from the new regulations. To prepare for the new procedure, the whole process should be analyzed by experienced experts who can prepare a clear path for your medical device registration in the EAEU.



")

")
")
