Regulatory affairs for biosimilars in Middle East North Africa (MENA) region are evolving as existing guidelines and regulatory frameworks are being updated. Since European Medicines Agency (EMA) introduced a legal pathway for biosimilars approval in 2004, several global agencies, e.g., WHO, MHRA, HC, have issued regulations and regulatory guidelines for biosimilars licensing. Saudi Arabia, Egypt, Lebanon, Jordan, Tunisia and Iraq have also published guidelines on biosimilar products. What follows is an overview of existing guidelines and regulatory framework for biosimilars in some of these countries.
Biosimilars’ development: regulatory affairs overview
Shortly put, “a biosimilar is a biological medicine highly similar to another already approved biological medicine”, the so-called “reference medicine”1.
Biological medicines’ natural variability and complex manufacturing process do not permit the exact replication of the active substance (“micro-heterogeneity”). The active substance is mostly a protein, has a way larger molecular structure than known chemical therapeutic entities, and is produced in a sophisticated process involving advanced technologies such as cell systems and recombinant DNA technology. The classical “generic concept” does not apply, and the related regulatory framework is not suitable for evaluating a candidate biosimilar.
Biosimilar development aims to have a highly similar product to the reference biological medicine, which already went through a comprehensive scientific assessment and obtained approval for the designated indication(s).
This biosimilarity should be demonstrated in terms of:
- Structure
- Biological activity
- Efficacy
- Safety
- Immunogenicity profile.
Only then, safety and efficacy experience gained from clinical trials with the reference medicine can be relied on, and unnecessary repetition of the full clinical program can be avoided. The below illustration compares the data requirements for approval of a biosimilar versus the reference medicine.
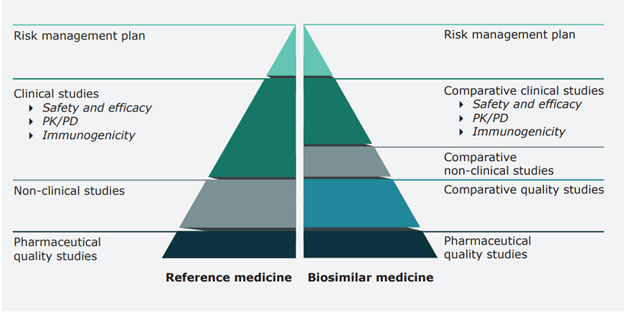
Figure 1. Comparison of data requirements for approval of a biosimilar vs. a reference medicine (EMA, 2020)
In practice, biosimilarity is proven by comprehensive comparability studies with reference medicine. As a result, a biosimilar may be used for the same indication. Extrapolation to other indications is possible as long as it is supported by all the scientific evidence generated in comparability studies (quality, non-clinical and clinical). Once approved and marketed, the biosimilar safety is surveilled through existing pharmacovigilance systems for medicines.
Biosimilars can improve patients’ access to quality biological medicines and bring several benefits to healthcare systems. However, it is also well-understood that the risk of insufficient quality, safety or efficacy is relatively higher than with other medicines, e.g., generics. Hence the importance of a solid biosimilar regulatory framework that can “strike a balance between regulatory standards, patient safety and feasibility of biosimilar development”[1].
Biosimilars’ regulatory affairs in the MENA region
All three MENA countries below have adopted biosimilars guidelines even though a complete and dedicated regulatory pathway may not be everywhere present.
Saudi Arabia: biosimilars’ regulatory overview
In general, the Saudi Food & Drug Authority (SFDA) regulatory framework governing drugs licensing applies to biosimilars as well. Regulations and guidelines extensively reference the US FDA regulations, with specificities accommodating for the local and regional, i.e., GCC, requirements.
Main information can be found in the following SFDA guidelines:
- Regulatory Framework for Drug Approval
- Guidance for Submission
- Guideline: GCC Data Requirements for Human Drugs Submission
- GCC Guidelines for Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products.
Specific guidelines on Biosimilars were published/adopted by the Saudi agency:
- Drug Master File Requirements for the Registration of Biosimilars (Dec. 2010)
- Guideline on Biosimilar Products Quality Considerations (Aug. 2017)
- The GCC Biosimilar Guideline (Aug. 2016), which is the only guideline in the region that provides detailed product-specific requirements, extensively based on the EMA and ICH relevant guidelines.
Of particular importance for preparing a biosimilar application for Saudi Arabia is the guideline that addresses the comparability topic. To assist applicants with the registration of biosimilars, the Drug Sector at the SFDA has published a document named “Guideline on Biosimilar Products – Quality considerations”, current version 1.0, addressing the quality aspects of the demonstration of biosimilar comparability and detailing the related requirements.
Regarding biosimilars interchangeability and as the topic continues to be discussed among all parties, SFDA recommends that changing from an innovator drug to a biosimilar drug that used that same innovator drug as its Reference Medicinal Product (RMP) for comparability, or vice versa, can be accepted after discussion and agreement between physician and patient. The same would apply for changing from a biosimilar drug to another similar biosimilar drug from a different manufacturer if they both used the same RMP for comparability purposes.
Changing from an innovator drug to another innovator drug for the same indication or from a biosimilar drug to another biosimilar drug that did not use the same innovator drug as an RMP for comparability is usually not acceptable. In extreme cases, physician and patient discussion, as well as hospital administration involvement in the decision, are mandatory.
Egypt: biosimilars’ regulatory overview
In Egypt, the guidelines for registration of Biosimilars were first issued in 2014 by the Egyptian Health Authority. These are now replaced by the current Egyptian Drug Authority’s (EDA) “Guideline for registration of Biosimilar products in Egypt” of March 2020. In the scope of the guideline are the well-characterized biologicals developed using biotechnology, including recombinant DNA technology. Vaccines and plasma-derived products and their recombinant analogues are excluded from the scope of these guidelines.
The guideline addresses the principles of the biosimilar approach, where the Applicant completes a comparability quality exercise and selected pre-clinical and clinical comparability studies to demonstrate biosimilarity of their product to a reference biological medicinal product, by mainly adopting the EMA biosimilars guidelines and referring to the USFDA’s scientific and quality considerations in demonstrating biosimilarity, to the WHO guidelines on evaluation of similar biotherapeutic products, and to the relevant ICH guidelines.
Furthermore, details of the assessment procedure by the EDA are provided, including steps and timelines and differentiation between locally manufactured and imported biosimilars. A further distinction is made depending on if the imported product – which must be approved and marketed in its country of origin – is manufactured in a “reference” country or not (see below figures taken from the EDA guideline). If not, a site inspection for GMP compliance will be required by EDA.
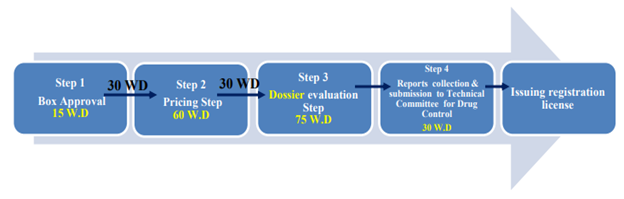
Figure 2. Flow chart representing the review steps for biosimilars imported from a “reference” country (EDA, 2021)
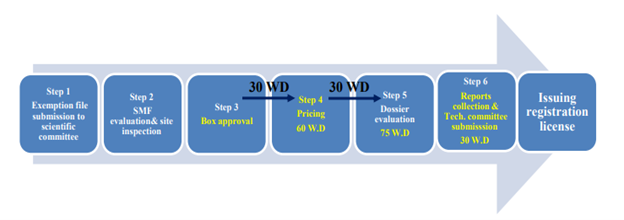
Figure 3. Flow chart representing the review steps for biosimilars imported from a “non-reference” country (EDA, 2021)
Since the Implementation of the Biosimilar Guidelines in Egypt and till May 2020, EDA had licensed 4 imported biosimilars and 10 candidate products were cleared to submit a MA (so-called “preliminary approval”). Also, 25 local products got preliminary approval to proceed with MA filing, part of them already having a MA under review.
There are yet no defined rules or established product lists in Egypt with regards to interchangeability, switching or substitution.
Jordan: biosimilars’ regulatory overview
The “guidelines for the registration of biosimilar drugs of 2015” are the Jordanian FDA’s Biosimilars guidelines, adopted on May 7, 2015. The document explicitly references in Article 12 the EMA biosimilar guidelines and explains that these are fully adopted, including their future revisions and updates. In other terms, Jordan has implemented the EMA model for comparability, addressing quality, non-clinical and clinical, as well as the product-specific requirements.
The Biosimilars registration pathway in Jordan provides information on the requirements for the Marketing Authorization Application (MAA) of a Biosimilar, including:
- An Application in eCTD format
- Module 1: complete, country-specific as per JFDA requirements
- Module 2: summaries
- Module 3: full, addition: the comparability quality part
- Module 4: comparability, non-clinical part (pharmacodynamics, toxicology)
- Module 5: comparability, clinical part (pharmacokinetics, pharmacodynamics, efficacy, safety)
- An RMP/Pharmacovigilance Plan addressing potential risks, including immunogenicity.
It also mandates Manufacturing site(s) accreditation as a prerequisite to product filing and approval. The accreditation includes the API manufacturing site and any site involved in the production of the finished product. An on-site GMP inspection by JFDA inspectors may be required. Exemptions are typically granted to those manufacturing sites that are based in a “reference” country, where the JFDA expects strict control of the manufacturing facility by the local competent authorities. In this case, foreign-issued certificates are accepted.
Reference countries and agencies are USA, EMA, Japan, Canada, UK, Germany, France, Belgium, Switzerland, Netherland, Sweden, Australia, Austria, Finland and Spain.
Regarding interchangeability, switching and substitution, the biosimilar guidelines indicate that the “decision to treat a new patient with either a reference product or its biosimilar medicine, or switch in an already treated patient between a reference product and its biosimilar should only be taken following the opinion of a qualified health professional”. It further mentions that automatic substitution cannot be applied.
Since the biosimilar guidelines of 2015 were published, JFDA announced in 2020 the approval of 6 products and rejection of one due to inadequate quality comparability and insufficient immunogenicity data. Overall, the JFDA approach in regulating biosimilars can be described as stringent and cautious. Nevertheless, products manufactured and marketed in “reference” countries are usually privileged.
In summary, biosimilars are widely valued for improving the patient’s access to critical biological treatments by offering an additional supply source and reducing the financial burden on healthcare systems. Regulatory standards and pathways for biosimilars exist now globally for more than a decade, and much experience has been gained from assessing and approving numerous products worldwide.
In the MENA region, countries like Saudi Arabia, Egypt and Jordan have adopted biosimilar guidelines that resemble the EMA, WHO and FDA guidelines and espouse their current understanding of biosimilars, which should ease the access to these market for foreign biosimilars developers. Moreover, as local Regulators tend to be still cautious and conservative in approaching and assessing biosimilars, the concept of the “reference” approval/country becomes of great importance: biosimilars are favoured when already approved by a stringent regulatory agency.
If you wish to get more information on this or other Regulatory affairs topics related to the MENA region, contact us!
[1] European Medicines Agency. Biosimilar Medicines Overview.
[2] Wolff-Holz, E. et al. (2019) „Evolution of the EU Biosimilar Framework: Past and Future“. BioDrugs 33:1 – 14.



")

")
")
